A large synthetic peptide and phosphopeptide reference library for mass spectrometry-based proteomics
We present a peptide library and data resource of >100,000 synthetic, unmodified peptides and their phosphorylated counterparts with known sequences and phosphorylation sites. Analysis of the library by mass spectrometry yielded a data set that we used to evaluate the merits of different search engines (Mascot and Andromeda) and fragmentation methods (beam- type collision-induced dissociation (HCD) and electron transfer dissociation (ETD)) for peptide identification. We also compared the sensitivities and accuracies of phosphorylation-site localization tools (Mascot Delta Score, PTM score and phosphoRS), and we characterized the chromatographic behavior of peptides in the library.
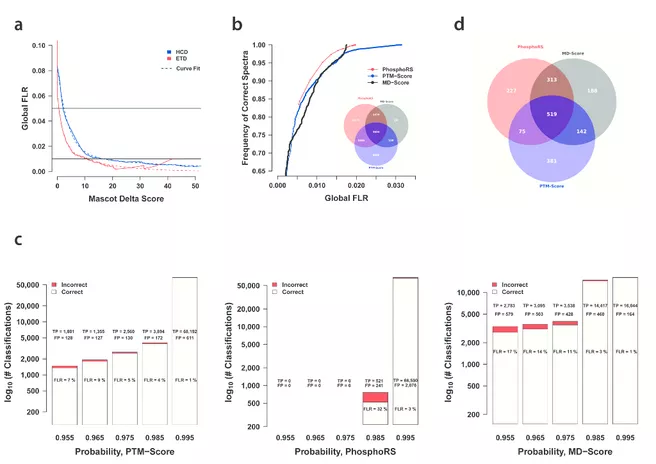
False Localization Rate (FLR) determination for phosphorylated peptides.
We found that HCD identified more peptides and phosphopeptides than did ETD, that phosphopeptides generally eluted later from reversed-phase columns and were easier to identify than unmodified peptides and that current computational tools for proteomics can still be substantially improved. These peptides and spectra will facilitate the development, evaluation and improvement of experimental and computational proteomic strategies, such as separation techniques and the prediction of retention times and fragmentation patterns.